Sample queries of DAMA on Essentia Proteomica server
Protein kinases
Query
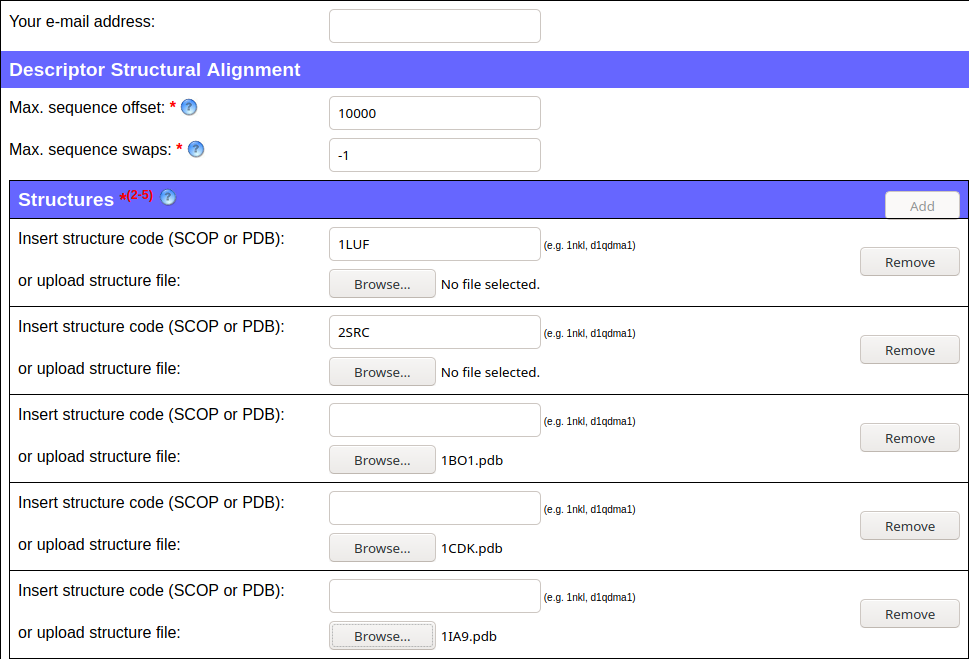
This example refers to proteins that have already been studied, where manually curated alignment has been prepared and precisely described [1]. As the reference structure for the alignment we propose to use Protein Kinase A (PKA; PDB: 1CDK), two typical kinases (TKs; PDB: 2SRC and 1LUF) and two atypical ones (AKs or ATKs; PDB: 1BO1 and 1IA9). There are features that should be aligned correctly in all structures mentioned above such as conserved histidine (at position 158 in 1CDK) and other structural elements that occur only in typical kinases, like helices called G, H and I.
To carry out the required computation:
- go to the DAMA website
- add slots for 5 input proteins
- input PDB ids or upload files from your local hard drive
- submit the job
In this case, the 1LUF and 2SRC structures are to be taken from PDB and the other three files were edited locally (removal of all chains but one) and uploaded from the local hard drive. The same would have been achieved if an additional chain letter had been added to structure ID in PDB, e.g. 1BO1A, 1CDKA or 1IA9A.
The screenshot below shows the query to compare these structures.
Computation
After successfully submitting the query user receives a message with a link to the computation results.

Although it may take several minutes to queue and execute a job, the link is active and redirects to an automatically refreshing page informing that the query is being processed.
Results
When the user's computation is done, the resulting page will show the following information:
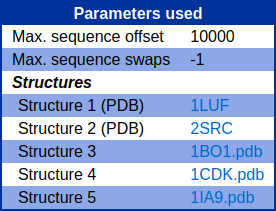
Parameters
The query parameters including max sequence offset, max sequence swaps as well as the compared structures, are displayed.
It should be noted that for structures downloaded from public databases, their names are shown in brackets next to structure numbers.
The structure identifier is a hyperlink to a PDB file.
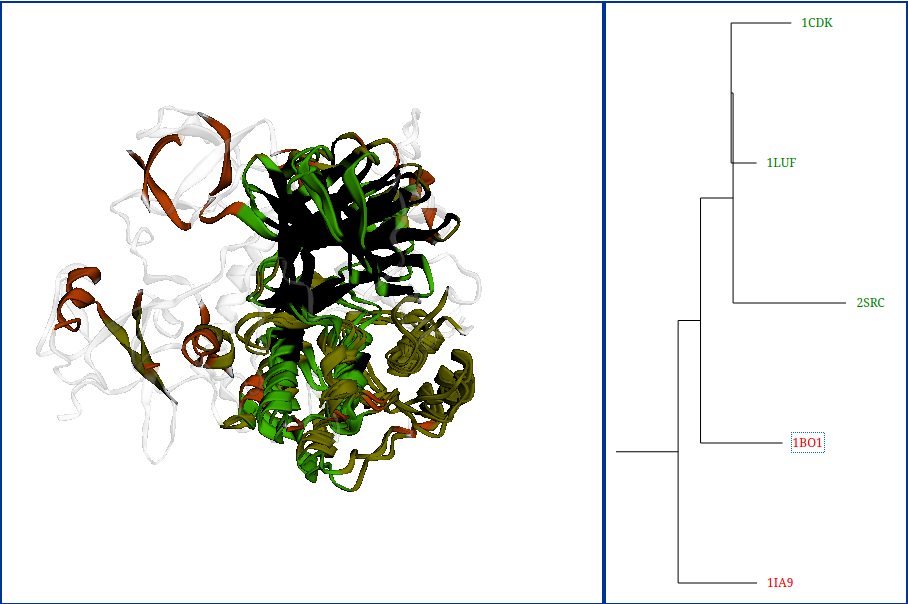
Superposition
Graphical representation of alignment in which compared structures are superimposed to minimize RMSD of the alignment residues.
Whole structures are presented, but only the aligned residues are highlighted.
Users are able to rotate the superimposed structures (LMB) and zoom in or zoom out (RMB) them.
An alignment tree shows the structural similarity between aligned structures.
Clicking on the structure identifiers shown in this section hides or shows structures in the section displaying structures
superimposition.
Hidden structures are marked in red, while shown are in green.
By default, the best alignment is displayed.
Scores
This table shows the five best-rated alignments.
Subsequent columns show:
- number of the aligned residues (N. AA)
- alignment score
- mean RMSD
- hyperlink to the alignment files
- hyperlink to the pdb file with superimposed structures
The "Show" button in the "Superposition" column displays the alignment superposition and the tree.
Results stored in the "Alignment" column are given as FASTA or PAL ("Ranges").
The former one is a well-known format listing sequences of the aligned structures with one-letter code,
presenting the aligned residues with capital letters, not aligned with lowercase and gaps as dashes.
The PAL format lists all structures pairs showing ranges of the aligned residues.
